
UCLA Health technical project manager Robert Smith joins for a discussion on the state of personalized medicine and computational medicine.
The goal of personal genomics is to move toward being able to interpret each person
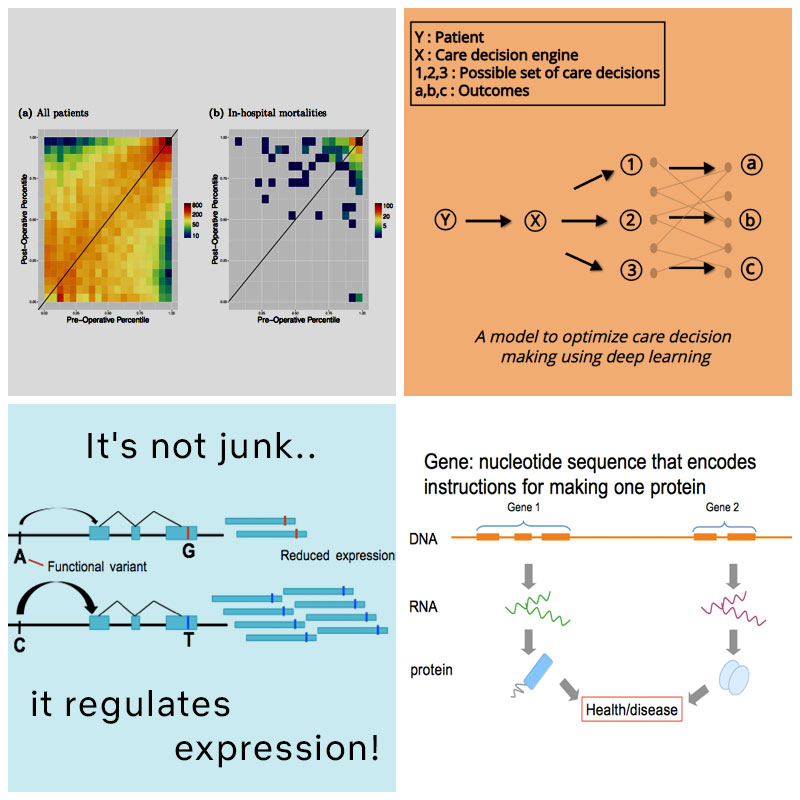
Predicting preoperative in-hospital mortality using readily-available electronic medical record (EMR) data can aid clinicians in accurately and rapidly determining surgical risk. While previous work has shown that the American Society of Anesthesiologists (ASA) Physical Status Classification is a useful, though subjective, feature for predicting surgical outcomes, obtaining this classification requires a clinician to review the patient’s medical records. Our goal here is to create an improved risk score using electronic medical records and demonstrate its utility in predicting in-hospital mortality without requiring clinician-derived ASA scores.
Although genome-wide association studies (GWAS) for prostate cancer (PrCa) have identified more than 100 risk regions, most of the risk genes at these regions remain largely unknown. Here we integrate the largest PrCa GWAS (N